Reproducible SLURM jobs from a Jupyter Notebook
For Jupyter notebook and Python lover, we can start automating our workflows by creating notebooks containing any number of pre-processing steps, batch scripts, monitoring commands and post-processing steps to be performed during and after job execution.
This can make HPC workflows more reproducible and shareable, and ready-made notebooks can make it easier, for example, for new reseacher students to get started.
In this post, instead of manage jobs via SSH terminal or open on demand web portal, we demo how to use Slurm Magics to do the interactive analysis and Slurm job management without leaving from Jupyter Notebook.
SLURM magics
- Slurm magic developed by National Energy Research Scientific Computing (NERSC)[1]
- The slurm magic command will interact with Slurm workload management, for short, it is Slurm command wrapper.
- Each command implement by fork or spawned new __subprocess then output is captured and show on notebook with UTF-8 decoding.
Using SLURM magics
![image.png]()
Assume, you connect to exascale.mahidol.ac.th portal, and create Jupyter Notebook server.
![image.png]()
In new jupyter notebook, we need to load IPython slurm extension:
1
2
| pip install git+https://github.com/NERSC/slurm-magic.git
|
From now on, we can interact with Slurm workload manager,without VPN SSH
1
2
3
4
5
6
7
| Available line magics:
%alias %alias_magic %autoawait %autocall %automagic %autosave %bookmark %cat %cd %clear %colors %conda %config %connect_info %cp %debug %dhist %dirs %doctest_mode %ed %edit %env %gui %hist %history %killbgscripts %ldir %less %lf %lk %ll %load %load_ext %loadpy %logoff %logon %logstart %logstate %logstop %ls %lsmagic %lx %macro %magic %man %matplotlib %mkdir %more %mv %notebook %page %pastebin %pdb %pdef %pdoc %pfile %pinfo %pinfo2 %pip %popd %pprint %precision %prun %psearch %psource %pushd %pwd %pycat %pylab %qtconsole %quickref %recall %rehashx %reload_ext %rep %rerun %reset %reset_selective %rm %rmdir %run %sacct %sacctmgr %salloc %sattach %save %sbatch %sbcast %sc %scancel %scontrol %sdiag %set_env %sinfo %slurm %smap %sprio %squeue %sreport %srun %sshare %sstat %store %strigger %sview %sx %system %tb %time %timeit %unalias %unload_ext %who %who_ls %whos %xdel %xmode
Available cell magics:
%%! %%HTML %%SVG %%bash %%capture %%debug %%file %%html %%javascript %%js %%latex %%markdown %%perl %%prun %%pypy %%python %%python2 %%python3 %%ruby %%sbatch %%script %%sh %%svg %%sx %%system %%time %%timeit %%writefile
Automagic is ON, % prefix IS NOT needed for line magics.
|
1
2
| import warnings
warnings.filterwarnings('ignore')
|
Submit GROMACS job and analysis results on the fly
To demo how to submit job for __ simulations of biological macromolecules__ GROMACS package example for Lysozyme[3] in water is used.
1
| !git clone https://github.com/snitgit/Slurm-jupyter-notebook.git
|
1
| cd Slurm-jupyter-notebook/
|
1
| /home/snit.san/slurm-magic/Slurm-jupyter-notebook
|
1
| /home/snit.san/slurm-magic/Slurm-jupyter-notebook/gromacs_job
|
| PARTITION | AVAIL | TIMELIMIT | NODES | STATE | NODELIST |
|---|
| 0 | batch* | up | 420-00:00: | 1 | mix | omega |
|---|
| 1 | batch* | up | 420-00:00: | 3 | idle | tensorcore,turing,zeta |
|---|
Use %sbatch to submit job on next cell
| JOBID | PARTITION | NAME | USER | ST | TIME | NODES | NODELIST(REASON) |
|---|
| 0 | 6599 | batch | sys-dash | snit.san | R | 1:13 | 1 | omega |
|---|
| 1 | 5890 | batch | bash | tantip.a | R | 9-02:06:59 | 1 | omega |
|---|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
| %%sbatch
#!/bin/bash -l
#SBATCH -A ict
#SBATCH -N 1
#SBATCH -t 01:05:00
#SBATCH -J gromacs
#SBATCH --gres=gpu:2
#SBATCH -w, --nodelist=zeta
# change temp or log to your folder
export SINGULARITY_TMPDIR=/home/snit.san/tmp
export CUDA_MPS_LOG_DIRECTORY=/home/snit.san/var/log/mvidia-mps
module use /shared/software/software/mulabs
module load hpcx-ompi
module load gromacs
gmx grompp -f npt.mdp -c start.gro -p topol.top -maxwarn 100
gmx mdrun -ntmpi 1 -ntomp 40 -v -pin on -nb gpu --pme gpu -noconfout -s topol.tpr -deffnm npt
|
1
| 'Submitted batch job 6611\n'
|
| JOBID | PARTITION | NAME | USER | ST | TIME | NODES | NODELIST(REASON) |
|---|
| 0 | 6599 | batch | sys-dash | snit.san | R | 44:26 | 1 | omega |
|---|
| 1 | 6611 | batch | gromacs | snit.san | R | 0:03 | 1 | zeta |
|---|
Gromacs utility can be used to extract information from the binary output files.
To run it, we write shell commands into a code cell containing the %%bash magic to let Jupyter execute a bash script. In our case, we extract time-dependent values of temperature, density and pressure from the simulation[4].
1
2
3
4
5
6
| %%bash
module use /shared/software/software/mulabs
module load gromacs/2021
echo "Temperature" | gmx energy -f npt.edr -o temperature.xvg
echo "Density" | gmx energy -f npt.edr -o density.xvg
echo "Pressure" | gmx energy -f npt.edr -o pressure.xvg
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
| Statistics over 161501 steps [ 0.0000 through 323.0000 ps ], 1 data sets
All statistics are over 16151 points
Energy Average Err.Est. RMSD Tot-Drift
-------------------------------------------------------------------------------
Temperature 300.024 0.069 1.66438 0.354538 (K)
Statistics over 162501 steps [ 0.0000 through 325.0000 ps ], 1 data sets
All statistics are over 16251 points
Energy Average Err.Est. RMSD Tot-Drift
-------------------------------------------------------------------------------
Density 1016.21 0.21 2.37206 -0.433522 (kg/m^3)
Statistics over 163501 steps [ 0.0000 through 327.0000 ps ], 1 data sets
All statistics are over 16351 points
Energy Average Err.Est. RMSD Tot-Drift
-------------------------------------------------------------------------------
Pressure 1.06924 0.18 140.482 0.193272 (bar)
INFO: Using cached SIF image
:-) GROMACS - gmx energy, 2021-dev-20210128-6a0b0c4-dirty-unknown (-:
GROMACS is written by:
Andrey Alekseenko Emile Apol Rossen Apostolov
Paul Bauer Herman J.C. Berendsen Par Bjelkmar
Christian Blau Viacheslav Bolnykh Kevin Boyd
Aldert van Buuren Rudi van Drunen Anton Feenstra
Gilles Gouaillardet Alan Gray Gerrit Groenhof
Anca Hamuraru Vincent Hindriksen M. Eric Irrgang
Aleksei Iupinov Christoph Junghans Joe Jordan
Dimitrios Karkoulis Peter Kasson Jiri Kraus
Carsten Kutzner Per Larsson Justin A. Lemkul
Viveca Lindahl Magnus Lundborg Erik Marklund
Pascal Merz Pieter Meulenhoff Teemu Murtola
Szilard Pall Sander Pronk Roland Schulz
Michael Shirts Alexey Shvetsov Alfons Sijbers
Peter Tieleman Jon Vincent Teemu Virolainen
Christian Wennberg Maarten Wolf Artem Zhmurov
and the project leaders:
Mark Abraham, Berk Hess, Erik Lindahl, and David van der Spoel
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2019, The GROMACS development team at
Uppsala University, Stockholm University and
the Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
GROMACS is free software; you can redistribute it and/or modify it
under the terms of the GNU Lesser General Public License
as published by the Free Software Foundation; either version 2.1
of the License, or (at your option) any later version.
GROMACS: gmx energy, version 2021-dev-20210128-6a0b0c4-dirty-unknown
Executable: /usr/local/gromacs/avx2_256/bin/gmx
Data prefix: /usr/local/gromacs/avx2_256
Working dir: /home/snit.san/slurm-magic/Slurm-jupyter-notebook/gromacs_job
Command line:
gmx energy -f npt.edr -o temperature.xvg
Opened npt.edr as single precision energy file
Select the terms you want from the following list by
selecting either (part of) the name or the number or a combination.
End your selection with an empty line or a zero.
-------------------------------------------------------------------
1 Bond 2 Angle 3 Proper-Dih. 4 Ryckaert-Bell.
5 LJ-14 6 Coulomb-14 7 LJ-(SR) 8 Disper.-corr.
9 Coulomb-(SR) 10 Coul.-recip. 11 Potential 12 Kinetic-En.
13 Total-Energy 14 Conserved-En. 15 Temperature 16 Pres.-DC
17 Pressure 18 Constr.-rmsd 19 Box-X 20 Box-Y
21 Box-Z 22 Volume 23 Density 24 pV
25 Enthalpy 26 Vir-XX 27 Vir-XY 28 Vir-XZ
29 Vir-YX 30 Vir-YY 31 Vir-YZ 32 Vir-ZX
33 Vir-ZY 34 Vir-ZZ 35 Pres-XX 36 Pres-XY
37 Pres-XZ 38 Pres-YX 39 Pres-YY 40 Pres-YZ
41 Pres-ZX 42 Pres-ZY 43 Pres-ZZ 44 #Surf*SurfTen
45 Box-Vel-XX 46 Box-Vel-YY 47 Box-Vel-ZZ 48 T-Protein
49 T-non-Protein 50 Lamb-Protein
51 Lamb-non-Protein
Back Off! I just backed up temperature.xvg to ./#temperature.xvg.2#
Last energy frame read 323 time 323.000
GROMACS reminds you: "Der Ball ist rund, das Spiel dauert 90 minuten, alles andere ist Theorie" (Lola rennt)
INFO: Using cached SIF image
:-) GROMACS - gmx energy, 2021-dev-20210128-6a0b0c4-dirty-unknown (-:
GROMACS is written by:
Andrey Alekseenko Emile Apol Rossen Apostolov
Paul Bauer Herman J.C. Berendsen Par Bjelkmar
Christian Blau Viacheslav Bolnykh Kevin Boyd
Aldert van Buuren Rudi van Drunen Anton Feenstra
Gilles Gouaillardet Alan Gray Gerrit Groenhof
Anca Hamuraru Vincent Hindriksen M. Eric Irrgang
Aleksei Iupinov Christoph Junghans Joe Jordan
Dimitrios Karkoulis Peter Kasson Jiri Kraus
Carsten Kutzner Per Larsson Justin A. Lemkul
Viveca Lindahl Magnus Lundborg Erik Marklund
Pascal Merz Pieter Meulenhoff Teemu Murtola
Szilard Pall Sander Pronk Roland Schulz
Michael Shirts Alexey Shvetsov Alfons Sijbers
Peter Tieleman Jon Vincent Teemu Virolainen
Christian Wennberg Maarten Wolf Artem Zhmurov
and the project leaders:
Mark Abraham, Berk Hess, Erik Lindahl, and David van der Spoel
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2019, The GROMACS development team at
Uppsala University, Stockholm University and
the Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
GROMACS is free software; you can redistribute it and/or modify it
under the terms of the GNU Lesser General Public License
as published by the Free Software Foundation; either version 2.1
of the License, or (at your option) any later version.
GROMACS: gmx energy, version 2021-dev-20210128-6a0b0c4-dirty-unknown
Executable: /usr/local/gromacs/avx2_256/bin/gmx
Data prefix: /usr/local/gromacs/avx2_256
Working dir: /home/snit.san/slurm-magic/Slurm-jupyter-notebook/gromacs_job
Command line:
gmx energy -f npt.edr -o density.xvg
Opened npt.edr as single precision energy file
Select the terms you want from the following list by
selecting either (part of) the name or the number or a combination.
End your selection with an empty line or a zero.
-------------------------------------------------------------------
1 Bond 2 Angle 3 Proper-Dih. 4 Ryckaert-Bell.
5 LJ-14 6 Coulomb-14 7 LJ-(SR) 8 Disper.-corr.
9 Coulomb-(SR) 10 Coul.-recip. 11 Potential 12 Kinetic-En.
13 Total-Energy 14 Conserved-En. 15 Temperature 16 Pres.-DC
17 Pressure 18 Constr.-rmsd 19 Box-X 20 Box-Y
21 Box-Z 22 Volume 23 Density 24 pV
25 Enthalpy 26 Vir-XX 27 Vir-XY 28 Vir-XZ
29 Vir-YX 30 Vir-YY 31 Vir-YZ 32 Vir-ZX
33 Vir-ZY 34 Vir-ZZ 35 Pres-XX 36 Pres-XY
37 Pres-XZ 38 Pres-YX 39 Pres-YY 40 Pres-YZ
41 Pres-ZX 42 Pres-ZY 43 Pres-ZZ 44 #Surf*SurfTen
45 Box-Vel-XX 46 Box-Vel-YY 47 Box-Vel-ZZ 48 T-Protein
49 T-non-Protein 50 Lamb-Protein
51 Lamb-non-Protein
Back Off! I just backed up density.xvg to ./#density.xvg.2#
Last energy frame read 325 time 325.000
GROMACS reminds you: "It's Calling Me to Break my Bonds, Again..." (Van der Graaf)
INFO: Using cached SIF image
:-) GROMACS - gmx energy, 2021-dev-20210128-6a0b0c4-dirty-unknown (-:
GROMACS is written by:
Andrey Alekseenko Emile Apol Rossen Apostolov
Paul Bauer Herman J.C. Berendsen Par Bjelkmar
Christian Blau Viacheslav Bolnykh Kevin Boyd
Aldert van Buuren Rudi van Drunen Anton Feenstra
Gilles Gouaillardet Alan Gray Gerrit Groenhof
Anca Hamuraru Vincent Hindriksen M. Eric Irrgang
Aleksei Iupinov Christoph Junghans Joe Jordan
Dimitrios Karkoulis Peter Kasson Jiri Kraus
Carsten Kutzner Per Larsson Justin A. Lemkul
Viveca Lindahl Magnus Lundborg Erik Marklund
Pascal Merz Pieter Meulenhoff Teemu Murtola
Szilard Pall Sander Pronk Roland Schulz
Michael Shirts Alexey Shvetsov Alfons Sijbers
Peter Tieleman Jon Vincent Teemu Virolainen
Christian Wennberg Maarten Wolf Artem Zhmurov
and the project leaders:
Mark Abraham, Berk Hess, Erik Lindahl, and David van der Spoel
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2019, The GROMACS development team at
Uppsala University, Stockholm University and
the Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
GROMACS is free software; you can redistribute it and/or modify it
under the terms of the GNU Lesser General Public License
as published by the Free Software Foundation; either version 2.1
of the License, or (at your option) any later version.
GROMACS: gmx energy, version 2021-dev-20210128-6a0b0c4-dirty-unknown
Executable: /usr/local/gromacs/avx2_256/bin/gmx
Data prefix: /usr/local/gromacs/avx2_256
Working dir: /home/snit.san/slurm-magic/Slurm-jupyter-notebook/gromacs_job
Command line:
gmx energy -f npt.edr -o pressure.xvg
Opened npt.edr as single precision energy file
Select the terms you want from the following list by
selecting either (part of) the name or the number or a combination.
End your selection with an empty line or a zero.
-------------------------------------------------------------------
1 Bond 2 Angle 3 Proper-Dih. 4 Ryckaert-Bell.
5 LJ-14 6 Coulomb-14 7 LJ-(SR) 8 Disper.-corr.
9 Coulomb-(SR) 10 Coul.-recip. 11 Potential 12 Kinetic-En.
13 Total-Energy 14 Conserved-En. 15 Temperature 16 Pres.-DC
17 Pressure 18 Constr.-rmsd 19 Box-X 20 Box-Y
21 Box-Z 22 Volume 23 Density 24 pV
25 Enthalpy 26 Vir-XX 27 Vir-XY 28 Vir-XZ
29 Vir-YX 30 Vir-YY 31 Vir-YZ 32 Vir-ZX
33 Vir-ZY 34 Vir-ZZ 35 Pres-XX 36 Pres-XY
37 Pres-XZ 38 Pres-YX 39 Pres-YY 40 Pres-YZ
41 Pres-ZX 42 Pres-ZY 43 Pres-ZZ 44 #Surf*SurfTen
45 Box-Vel-XX 46 Box-Vel-YY 47 Box-Vel-ZZ 48 T-Protein
49 T-non-Protein 50 Lamb-Protein
51 Lamb-non-Protein
Back Off! I just backed up pressure.xvg to ./#pressure.xvg.2#
Last energy frame read 327 time 327.000
GROMACS reminds you: "If you want to destroy my sweater, hold this thread as I walk away." (Weezer)
|
define a function to extract data from the processed Gromacs xvg files
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
| def get_prop(prop):
"""Extract system property (Temperature, Pressure, Potential, or Density)
from a GROMACS xvg file. Returns lists of time and property."""
x = []
y = []
f_prop = open("%s.xvg" % prop, 'r')
for line in f_prop:
if line[0] == '#' or line[0] == '@':
continue
content = line.split()
x.append(float(content[0]))
y.append(float(content[1]))
f_prop.close()
return x,y
|
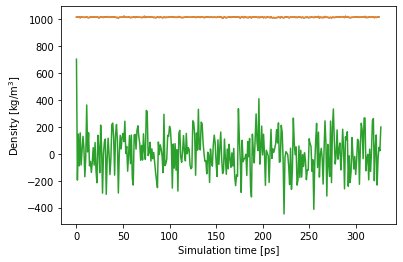
Having got data column from gromacs, we shoud diplay graph on Notebook using matplotlib.
1
2
3
4
5
6
7
8
9
10
11
12
| import matplotlib.pyplot as plt
%matplotlib inline
time,dens = get_prop("density")
plt.plot(time,dens)
plt.xlabel('Simulation time [ps]')
plt.ylabel('Density [kg/m$^3$]')
plt.plot(time,dens)
time,pres = get_prop("pressure")
plt.plot(time,pres)
|
1
| [<matplotlib.lines.Line2D at 0x7f3e490ecfd0>]
|
![Jupyter Notebook Plot]()
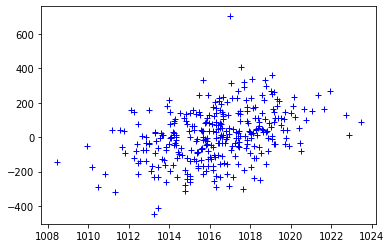
1
| plt.plot(dens,pres[:len(dens)],'b+')
|
1
| [<matplotlib.lines.Line2D at 0x7f3e49066dc0>]
|
![Jupyter Notebook Plot]()
References: 1. Slurm-magin https://github.com/NERSC/slurm-magic 2. Using Jupyter Notebooks to manage SLURM jobs https://www.kth.se/blogs/pdc/2019/01/using-jupyter-notebooks-to-manage-slurm-jobs/
1
2
3
4
5
| 3. GROMACS tutorial
http://www.mdtutorials.com/gmx/lysozyme/index.html
4. Using Jupyter Notebooks to manage SLURM jobs
https://www.kth.se/blogs/pdc/2019/01/using-jupyter-notebooks-to-manage-slurm-jobs/
|